5. Module 5¶
Module 5: Translation: The need for an Open Reading Frame
| Authors: | Carina Endres Howell (Lock Haven University of Pennsylvania)
and Leocadia Paliulis (Bucknell University) |
|---|---|
| Last Update: | Feb 24, 2020 |
| Version: | 0.0.1 |
5.1. Investigation 1: Examining Open Reading Frames in tra¶
Learning Objectives
- Use start and stop codons to identify open reading frames by looking at the Base Position track in the genome browser.
5.1.1. Introduction: review of reading frames¶
In this exploration, we will continue to focus on the transformer gene (referred to as tra-RA or just tra), and will learn about how the tra mRNA is translated into a string of amino acids.
Given that DNA is double-stranded, and that the genetic code is based on triplets (3 consecutive bases), there are six possible reading frames. One can determine a reading frame by dividing the sequence of nucleotides in DNA or RNA into a set of consecutive, non-overlapping triplets. There are three possible reading frames (read 5’ → 3’) in the forward direction on the top strand of DNA, and three (read 5’ → 3’) in the reverse direction on the complementary bottom strand of the same DNA molecule. Hence, there are six possible reading frames for each gene (see illustration in Module 1).
Once it is determined in which direction a particular gene is transcribed (for review see Module 2 and 3 on transcription), there remain three choices for the reading frame. To determine which of these reading frames is used during translation, evidence such as the presence of an initiation codon and the absence of stop codons is used. As you learned in Module 1, the initiation codon is ATG in the coding DNA strand (AUG in the mRNA) and specifies the amino acid methionine. Additional triplets code for the other 19 amino acids, and three triplets are stop codons, causing termination of translation. These stop codons are TAA, TAG and TGA in DNA, or UAA, UAG and UGA when found in mRNA). An Open Reading Frame is a string of consecutive codons that is uninterrupted by stop codons. Every mRNA contains one ORF that is translated by the ribosome from start codon to stop codon.
5.1.2. Investigate reading frames for the tra gene¶
- Go to the UCSC Genome Brower Mirror site at
http://gander.wustl.edu/ and follow the instructions given in
Module 1 to open
contig1of Drosophila melanogaster using theJuly 2014 (Gene)assembly. - The screen below will appear (Figure 5.1). As you will remember, this section of DNA is 11,000 base pairs long and is a small part of the left arm of chromosome 3, which is about 28,100,000 bp long.
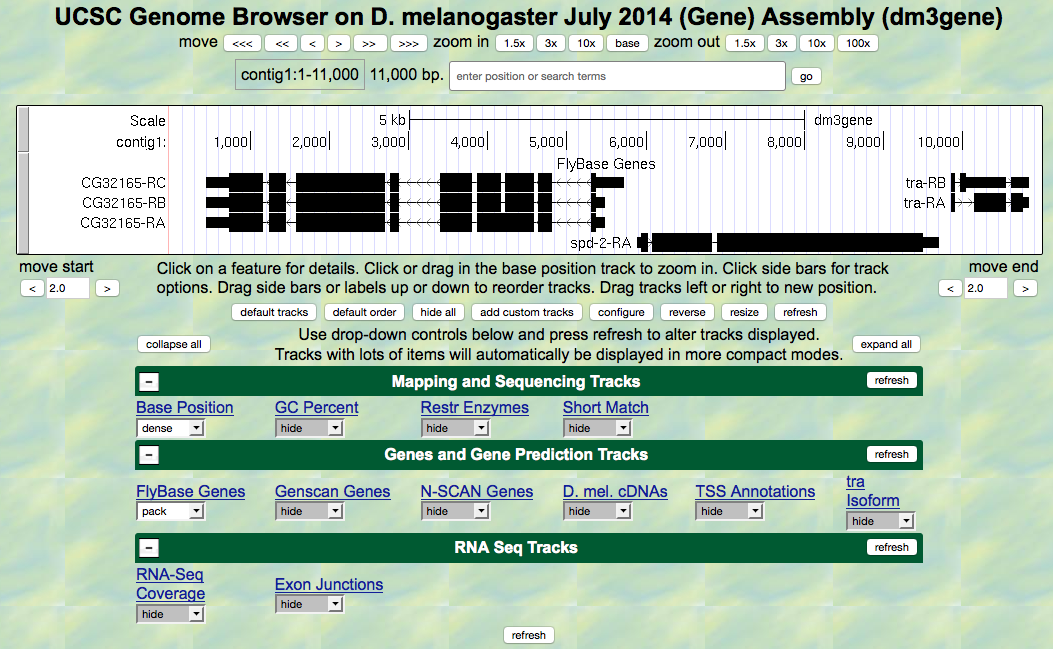
Figure 5.1. View of the region of the tra gene on Chromosome 3.
- Zoom in to view only the first exon of the tra-RA gene
by entering
contig1:9,840-9,920in the “enter position or search terms” text box and hitting thegobutton. - Open only the tracks that will provide information for this
investigation. Set the Base Position track to
full, and clickrefresh. Now we can see the amino acid tracks as well, giving the results from conceptual translation. - There are three possible reading frames for this transcript in the forward direction, here indicated by the numbers 1, 2 and 3 (red arrow) (Figure 5.2).
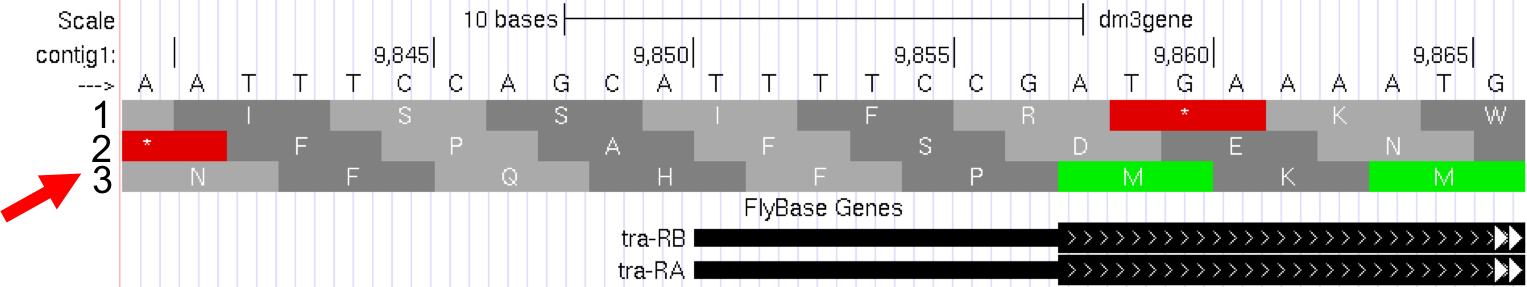
Figure 5.2. Three possible reading frames for the tra gene in the forward direction.
- Three reading frames are possible in the forward direction, as one
could start translating with the first base, the second
base or the third base. (Starting with the fourth base is
equivalent to starting with the first base, only missing one
codon.) The DNA “top strand” is read from left to right
(indicated by the arrow in the browser, right under the word
“contig1” that looks like this
--->). If you click on that arrow, the three reading frames in the reverse direction will appear, as the “bottom strand” is read from right to left. As you learned in Module 1, genes have directionality. The tra gene is read from the “top strand” (left to right). - Notice that the third reading frame has a green M codon (methionine) at the location where the thick black rectangle indicates the first coding exon or CDS (Coding DNA Sequence) of tra-RA mRNA. Remember that the codon for methionine (ATG in DNA) is the start signal, the first codon used in translation. This gives us our first piece of evidence that reading frame 3 is the one used in translation of the first CDS of the tra gene. For simplicity let’s call this first CDS “CDS1” to distinguish it from other CDS’s in the tra gene. Note that there is a stretch of RNA transcript upstream (to the left) of the ATG; this is the 5’UTR (5’ untranslated region), found at the 5’ end of all eukaryotic mRNAs.
- Next carefully examine reading frame 2. Notice that in this reading frame there is no codon for methionine (no start codon) in the region that maps to the first exon. This gives us evidence that reading frame 2 is probably not being used during translation of CDS1 of the tra gene.
- Finally, look at reading frame 1. Notice that there is a stop codon at the beginning of that reading frame (indicated by a red box with an asterisk in it). This evidence indicates that reading frame 1 probably is not being used during translation of CDS1 of the tra gene.
- Let’s move on to looking at the reading frames for exon 2 of tra-RA.
Zoom in to view only the second exon of the tra gene by jumping to
contig1:10,120-10,570using the “enter position or search terms” text box. Remember that both the RNA-Seq data and the cDNA data have been used to map the positions of exons.
Question 1
First examine reading frame 1. Are there any stop codons in the reported exon? If there are early stop codons, do you think this is the reading frame used during translation?
Question 2
Examine reading frame 2. Are there any stop codons in this reading frame within the exon?
Question 3
Examine reading frame 3. Are there any stop codons in the reported exon?
Question 4
Using the evidence above, which reading frame maintains an Open Reading Frame (ORF) across exon 2 of tra-RA? Is this the same reading frame as that used for exon 1?
- Finally, take a look at exon three (
contig1:10,600-10,850). We anticipate that since this is the last CDS there will be one or more stop codons, one of which will mark the site of translation termination. The 3’UTR (3’ untranslated region) extends downstream from this point to the site of poly(A) addition where the last exon ends (see Module 3). Here all three reading frames have an ORF followed by one or more stop codons in the exon.
Note
How can we figure out which reading frame is correct? We will investigate this in the next section by looking specifically at the splice junction.
5.2. Investigation 2: Construct the gene model for tra-RA¶
Learning Objectives
- Identify how to assemble exons to maintain the open reading frame (ORF) for a given gene.
- Define the phases of the splice donor and acceptor sites and how they impact the maintenance of the ORF.
- Identify start and stop codons of an assembled ORF.
- Use the coordinates of the start and stop codons and splice sites to generate a model of the coding region of a gene isoform.
We can combine what we know about reading frames with what we know about splicing to learn exactly how tra-RA is put together. We’ll note where the start codon, splice sites, and stop codon are so we can construct a gene model. Then, in Module 6, we’ll use these same types of information to solve some mysteries about tra-RB.
- Using the same Genome Browser page, reset the Browser by clicking on
hide all. Then open the tracks that will provide the information we want for Investigation 2:- Base Position:
full- Note that you will not be able to see the DNA sequence or amino acid tracks until you zoom in.
- FlyBase Genes:
pack - RNA-Seq Coverage:
full- You will see blue and red histograms representing the RNA-Seq data (indicating the amount of mRNA synthesized) in females and males, respectively.
- We will focus on the blue histogram (Adult Females)
again. As we did in Module 3,
let’s customize the RNA-Seq track:
- Click on the
RNA-Seq Coveragelabel under the RNA-Seq Tracks green bar found in the bottom section of the page. - Set the “Data view scaling” field to
use vertical viewing range setting - Set the “max” “Vertical Viewing range” to
37 - Under the “List sub-tracks” section, unselect the
Adult Malestrack
- Click on the
- Exon junctions:
full- These rectangular boxes joined by a thin black line will help us identify the exon-intron boundaries.
- Base Position:
5.2.1. Identify the start codon¶
- Let’s find the start codon for tra-RA. Zoom in on where the FlyBase Genes track shows that the translation starts (where the tracked black box gets thicker for the tra-RA isoform) as seen in (Figure 5.3)
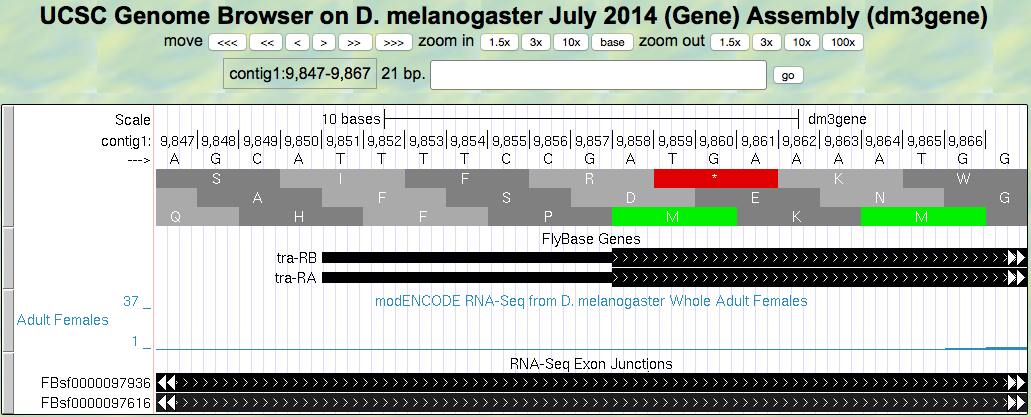
Figure 5.3. Translation start region of the tra gene.
Question 5
Give the coordinates for the entire start codon for tra-RA (start codon coordinates should be three consecutive numbers, for example: nucleotides 212–214).
Question 6
Which reading frame should we follow along to see the predicted amino acid sequence of tra-RA?
Question 7
Zoom out to see the entire exon. Are there any stop codons in this reading frame in the first exon?
5.2.2. Identify the splice sites for intron 1¶
- Now zoom in and find the last base of the first exon for tra-RA using your RNA-Seq data and Exon Junctions data (Figure 5.4).
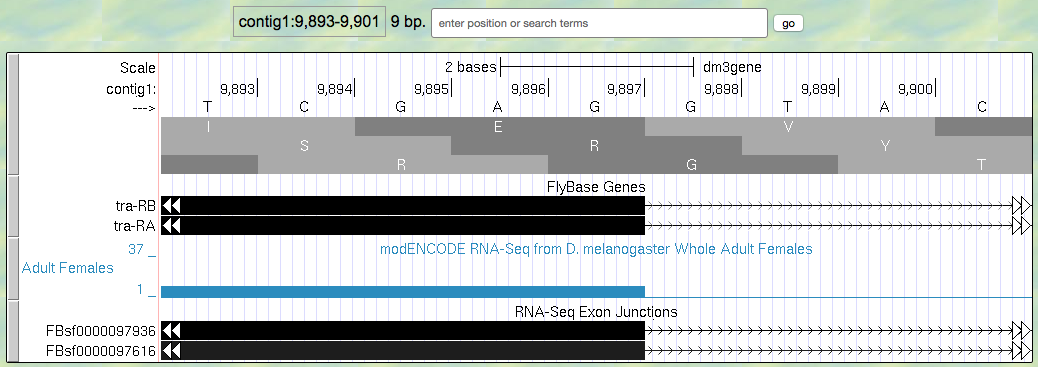
Figure 5.4. Region at the end of Exon 1 of the tra gene.
Question 8
Give the coordinates for the very last base of the first exon.
So that we can follow the polypeptide through until we identify the stop codon, we need to figure out which reading frame we should follow in the second exon. This is not as easy as you might think, because eukaryotes don’t always read in the same reading frame when looking at the genome. You saw an example of this previously in Module 1. Sometimes we can infer the correct reading frame given the pattern of start and stop codons within the region of the exon, identified by RNA-Seq data. But that sort of information does not always give a definitive answer — there may be more than one possible reading frame for a given exon. To figure out which reading frame is being translated at exon 2, we need to check the end of the first exon to see how many bases of the last codon are present before the 5’ splice site consensus sequence. To do this, look closely at reading frame 3, just before the splice site (Figure 5.5).
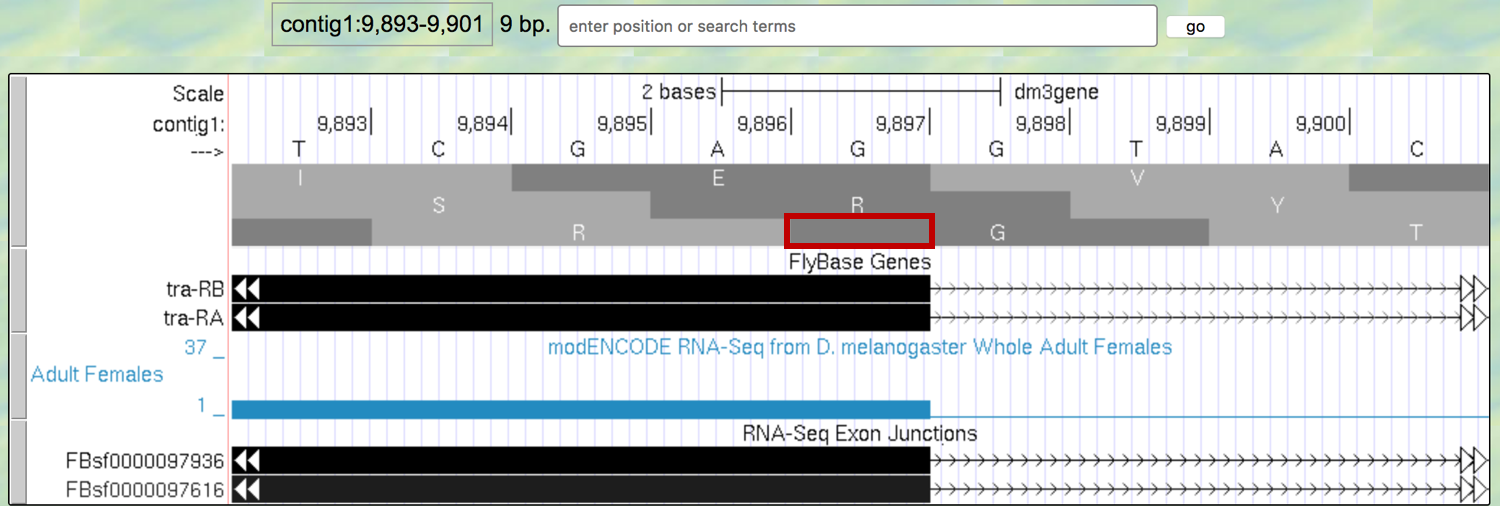
Figure 5.5. An extra nucleotide between the last complete codon and the splice donor site for Exon 1 of the tra gene.
Note that the splice site cuts off the last codon of the first exon after just one base (as indicated by the red box in Figure 5.5). Therefore, we would say this exon has a “phase 1” end because there is a partial codon at the end of the exon that is 1 base long.
Note
If there were a fully completed codon before the splice site, it would be in phase 0, and if there were two bases before the splice site, it would be in phase 2.
For this exon with a phase 1 end we will need two more bases from the next exon to complete the codon. Knowing this we can identify the reading frame that will be used in the second exon. Navigate to the 3’ splice site of intron 1 (i.e., the location where the first intron ends and the second exon begins; Figure 5.6). To review splicing and the concept of phase, watch the Splicing and Phase video.

Figure 5.6. Region at the beginning of Exon 2 of the tra gene
Question 9
Knowing that exon 1 ends with a partial codon of 1 base, what reading frame is being used in the second exon?
Question 10
Based on the evidence you see in the browser, give the coordinates for the first base of the second exon of tra-RA.
Question 11
Do you observe an appropriate splice acceptor site just upstream within the intron?
Now we will be using reading frame 2, because, after the splice site, there are two bases left in the codon. These two bases plus the one base left from the first exon make a complete codon.
- Next, zoom out and look at reading frame 2 for all of exon 2 of tra-RA. You can see that there are no stop codons in this reading frame, which lends support to our conclusion that this is the proper reading frame.
5.2.3. Identify the splice sites for intron 2¶
- Now, let’s do the same for the 5’ splice site of intron 2 for tra-RA. Zoom in on that splice site (Figure 5.7).
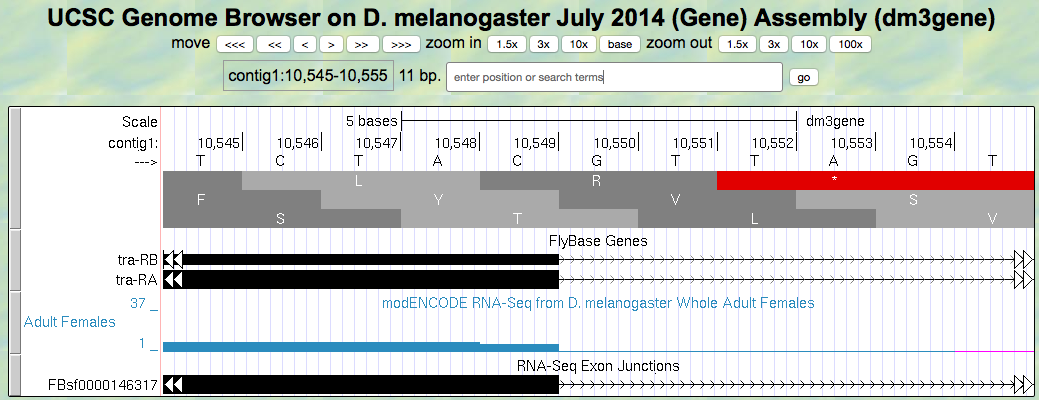
Figure 5.7. Region at the end of Exon 2 of the tra gene.
Question 12
Give the coordinate of the base prior to the 5’ splice site of intron 2.
Question 13
How many bases are left in the codon before the splice site, i.e. is this phase 0, phase 1, or phase 2?
6. Navigate to the start of the final exon (Figure 5.8).
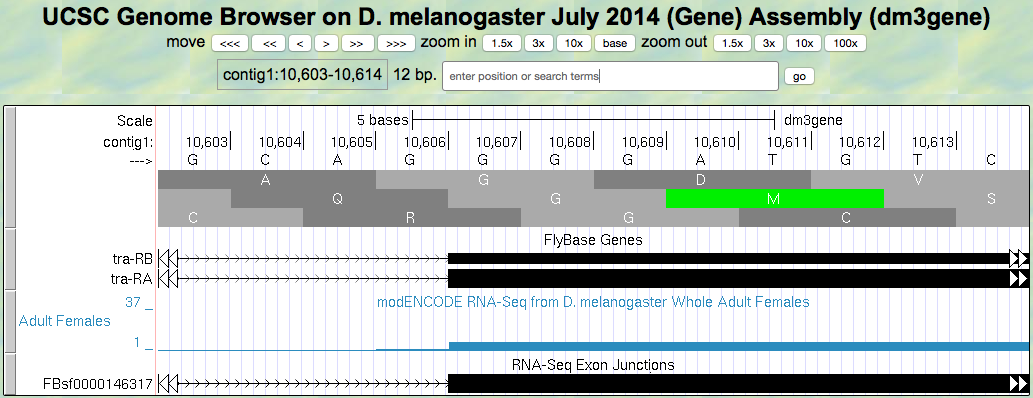
Figure 5.8. Region at the beginning of Exon 3 of the tra gene.
Question 14
Locate the 3’ splice site of Intron 2. Give the coordinate of the first base in exon 3 for tra-RA.
Question 15
Which reading frame is being translated in the final exon?
5.2.4. Identify the stop codon¶
- Now locate the first stop codon in the translated reading frame. Stop codons are shown as red boxes with asterisks (red arrows) as shown in Figure 5.9.
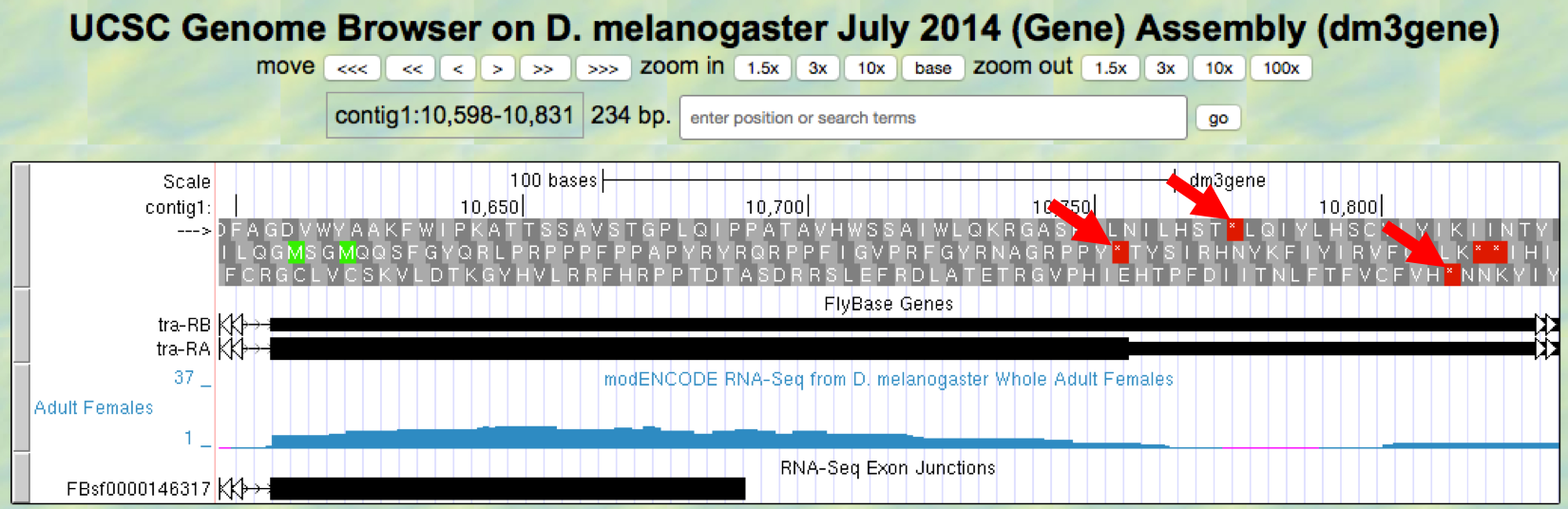
Figure 5.9. Region at the end of Exon 3 of the tra gene.
Question 16
Give the coordinates for the bases in the stop codon.
5.2.5. Construct the complete gene model¶
Let’s consolidate all the data we found above in one place:
Question 17
Gene model for tra-RA:
- Coordinate for start of translation: ______________
- Coordinate for last base of exon 1: ______________
- Coordinate for first base of exon 2: ______________
- Coordinate for last base of exon 2: ______________
- Coordinate for first base of exon 3: ______________
- Stop codon coordinates: ___________________________
Take the coordinate information above to draw a map of tra-RA using rectangles to represent exons and connecting lines to represent introns. Label the ends of the exons with the appropriate coordinates and indicate the transcription start site for the tra-RA initial transcript. Below this map, provide a map of the processed mRNA after intron removal. Below this map, indicate the regions that are translated into a protein. Give precise coordinates. Color coding may be helpful.
In Module 6, we will compare this model of tra-RA with a model of tra-RB.
Summary Question 1
To cement your knowledge of gene structure, you could construct a similar map of the spd-2 gene. How many exons does this gene have? How many introns? How many isoforms? Use the same approach to determine the coordinates for the exons, and the coordinates for the coding region (another name for the region that is translated).