Fragile X Syndrome
This is Sam. When he was a still a baby, his pediatrician noticed he had developmental delays. Sam continued to be behind his peers in terms of his emotional and intellectual development. He reacted negatively to changes in routine as well. Doctors performed a variety of psychological and medical tests on Sam, trying to determine the cause of his symptoms. The diagnoses ranged from hydrocephalus (excess cerebrospinal fluid in the brain) to autism. Sam had a male cousin with many of the same symptoms, suggesting a possible genetic link. When he was 5 1/2 years old, Sam was correctly diagnosed with Fragile X Syndrome (FXS).
According to the National Institutes of Health, FXS occurs in approximately 1/4000 males and 1/8000 females (Fragile X Syndrome, 2016).
Individuals with FXS often display psychological symptoms such as:
- mild to moderate cognitive impairment
- attention deficit-hyperactivity (ADHD)
- social anxiety
Approximately 5% of diagnosed cases of autism result from FXS.
Physical symptoms can commonly include:
- flexible joints
- macroorchidism (enlarged testes)
- an elongated face
- prominent chin
- large ears
With a diagnosis in hand, Sam’s parents were able to get him special services at school. Sam graduated from high school in 1993.

Sam is now 41 years old. He works at a local shopping mall and lives at home with his parents. Sam enjoys playing keyboard, listening to music, and watching baseball games.

The disorder is called fragile X syndrome because a region of the X chromosome can appear unbanded, constricted, and fragile when cells from affected individuals are cultured under appropriate conditions (Sutherland, 1977). (Click here to learn more.)
Folate-deficient medium is required to reveal the fragile site, and it does not always appear, even when the patient has demonstrable FXS (Sutherland, 1977; Hecht and Sutherland, 1985; Dewald, Buckley, Spurbeck, and Jalal, 1992). This and other late-replicating fragile sites throughout the genome are sites of chromosomal breakage and rearrangement (reviewed in Schwartz, Zlotorynski, and Kerem, 2006).
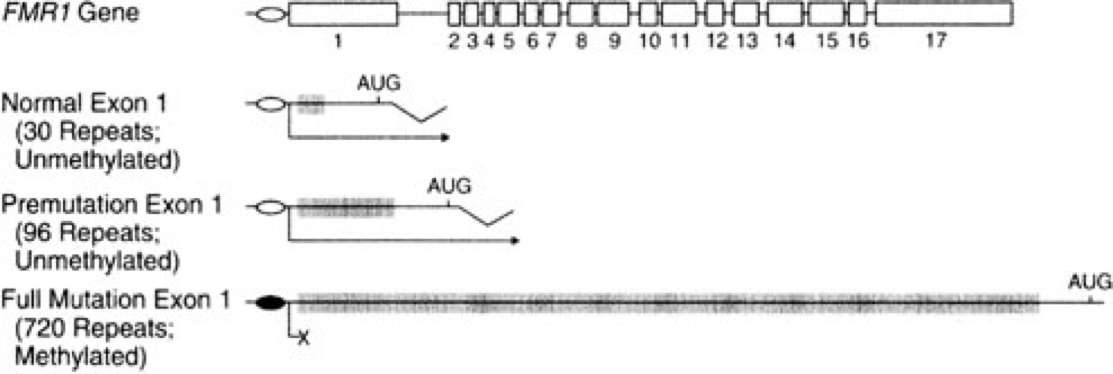
Today we know that FXS results from mutations in the FMR1 gene (short for Fragile X Mental Retardation; Verkerk et al., 1991). FMR1 is located on the X chromosome at genetic locus Xq27.3, the very region that appears unbanded and fragile in those characteristic chromosome preparations.
Deficits in FMR1 cause changes in the neurons of the central nervous system. We will explore these changes after a complete exploration of FXS genetics.

X-linked inheritance
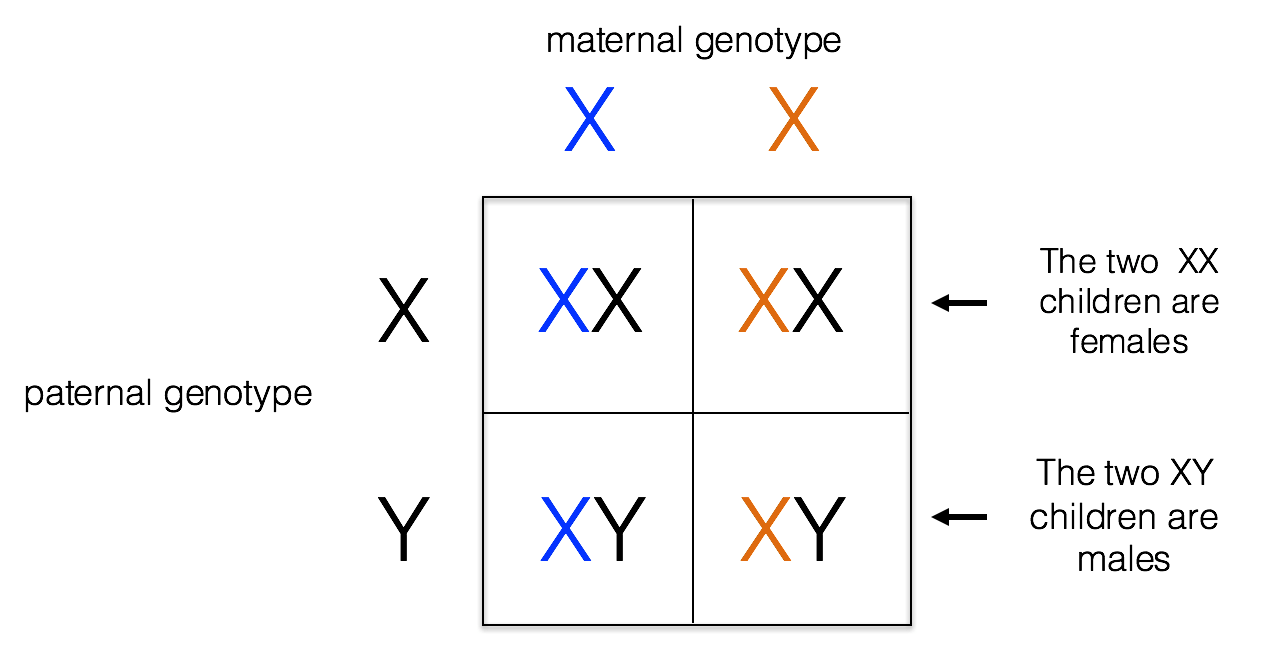
The X chromosome is one of two sex chromosomes in humans. It carries around 2000 genes. Females have two X chromosomes, while males have an X and a Y chromosome. The Y chromosome is necessary for male development. Because males are hemizygous for the X chromosome (having just one copy), alleles on the X chromosome are always expressed in males, whether they are formally dominant or recessive. Males inherit their X chromosome from their mother, as shown below.
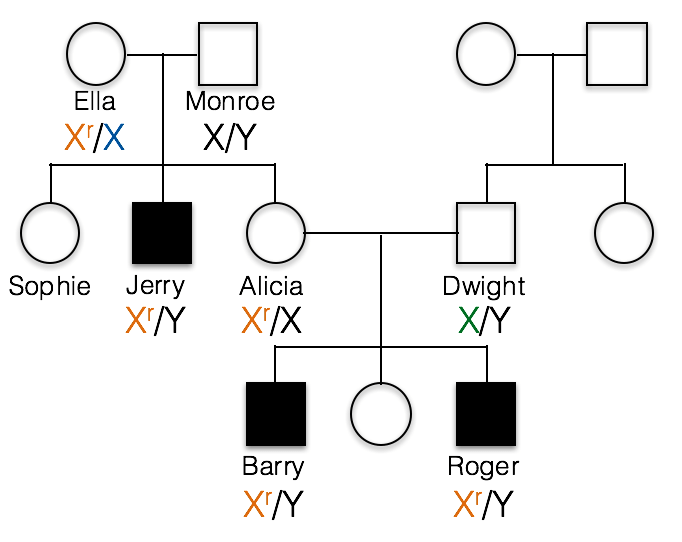
This pedigree shows the inheritance of a typical X-linked recessive trait. Females are designated by circles and males by squares. Individuals showing the recessive phenotype are shaded in. Jerry’s genotype is shown. He carries the recessive allele on his X chromosome, designated Xr.
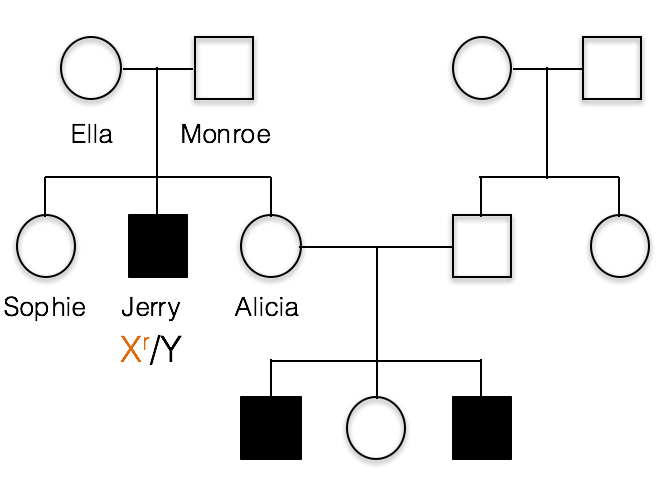
1(1). Which of the following best represents the genotypes of Jerry’s parents, Ella and Monroe?
A. incorrect; all of the children resulting from a cross of X/X with X/Y would have a normal phenotype.
B. incorrect; If Monroe had the recessive allele, the XrY genotype would result in a recessive phenotype. This is because males are hemizygous and Monroe has no dominant allele to hide the recessive allele.
C. Ella is heterozygous, Xr/X, and Monroe is X/Y as shown below.
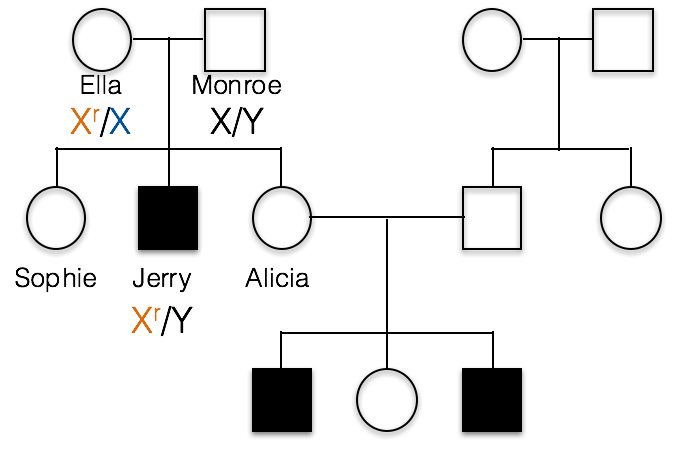
1(2) What would be the correct genotype for Alicia?
B. incorrect; Alicia must have at least one recessive Xr allele, otherwise she could not pass the trait on to her sons as she has done in this family.
C. incorrect; if Alicia were homozygous Xr/Xr, then she would have a recessive phenotype.
A. Alicia is Xr/X. We know Alicia carries the recessive allele because her sons Barry and Roger inherited this recessive allele from her and their Y chromosome from their father, Dwight.
2. This is the pedigree for Sam’s family. Like most X-linked disorders, only males appear to have symptoms. Sam’s first cousin (*) has FXS, as does a more distant cousin (#).
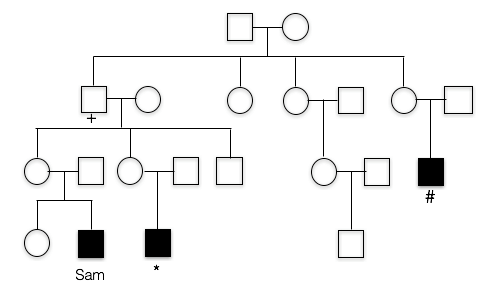
Does this trait appear to be dominant or recessive?
The trait does not appear in the parents of the unaffected children. Also note that those affected are all male, which is characteristics of many recessive, X-linked traits.
dominant: the trait does not appear in Sam’s mother, nor does it appear in Sam’s cousin’s mother, so it does not appear to be dominant.
3. What is unusual about Sam’s maternal grandfather(+)?
B. Yes, Sam’s grandfather doesn’t appear to have FXS (in fact, he had no phenotype whatsoever), despite the fact that he must have passed the mutated allele to his daughters (Sam’s mother and Sam’s aunt). Sam’s affected cousins show that FXS runs in this family.
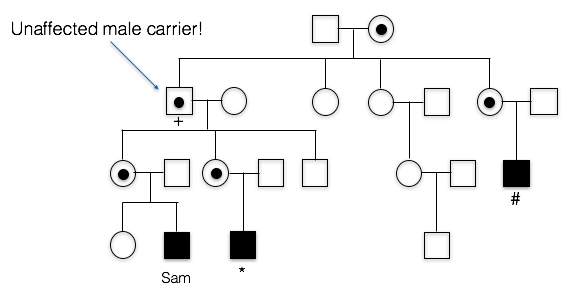
A. Incorrect. We know that the trait is running in Sam’s mother’s family. Since his first cousin as well as his more distant cousin have the trait as well, this means the trait probably originally came from Sam’s great grandmother, since she is the relative they all have in common. This means the FXS allele passed from Sam’s grandfather to Sam’s mother, to Sam. Sam’s grandfather doesn’t appear to have the X-linked trait, however.
C. Incorrect. Sam’s Grandfather did not have FXS. In fact, he graduated from college and showed no cognitive impairment whatsoever.
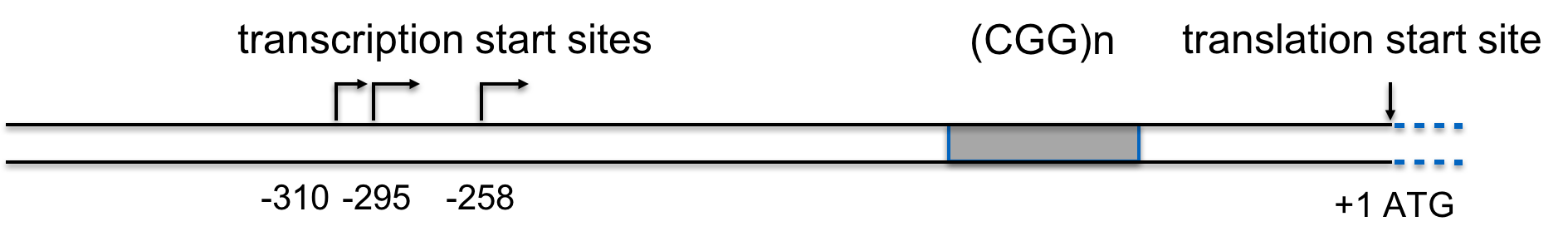
Sequencing the FMR1 gene provided some insights into the nature of the mutation causing FXS. In the partial sequence below, the three possible transcription start sites are indicated (TSS 3, TSS 2, and TSS 1), along with the start site for protein synthesis (+1 Translation Start)(Beilina et al., 2004). Click here for an interactive review of transcription and translation. The sequence that is underlined is a CGG trinucleotide repeated 16 times. Researchers have discovered that people with FXS contain many more copies of the (CGG)n sequence than unaffected individuals (Beilina et al., 2004; Kremer et al., 1991).

4. Based on the data, what kind of mutation results in most cases of FXS?
A. Incorrect. The mutation described involves the addition of many nucleotides, specifically the trinucleotide CGG repeat, rather than a single nucleotide. Furthermore, the nucleotide changes are 5’ to the translation start site and would not affect the protein in the same way that a missions mutation would. While point mutations in the FMR1 gene do occur (reviewed in Santoro, Bray, and Warren, 2012), these represent a minor percentage of mutations in all FXS cases.
B. Incorrect. The data indicate the addition of (CGG)n repeats in this region of the FMR1 gene occur in FXS patients. While deletions in the FMR1 gene do occur (reviewed in Santoro, Bray, and Warren, 2012), these represent a minor percentage of all FXS cases and were not the cause of Sam’s FXS.
C. Correct. The most common mutations in the FMR1 gene linked to FXS result from the expansion of a (CGG)n triplet repeat (Beilina et al., 2004; Kremer et al., 1991).
Triplet repeat expansions in other genes located on a variety of chromosomes have been linked to many human diseases. These diseases include Huntington’s Disease, Friedreich’s ataxia, and myotonic dystrophy (Suhl and Warren, 2015).
Individuals having between 55 and 200 copies of the (CGG)n repeat are said to have a premutation (Brasa et al., 2016; Hagerman and Hagerman, 2013). Males carrying more than 200 copies of the repeat will display FXS. The phenotype is quite variable in women, who have two X chromosomes. One of these X chromosomes is normally inactivated randomly as a means of achieving dosage compensation. Click here to learn more about other consequences of the (CGG)n expansion.
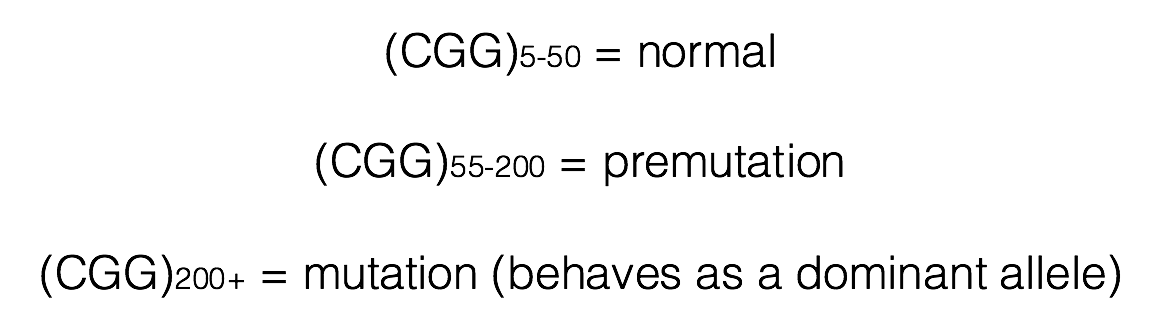
Males carrying the premutation may have Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS), a neurodegenerative disorder that typically presents over 50 years of age (FXTAS, 2015). In females, inheritance of the premutation can cause primary ovarian insufficiency (FXPOI; Fortuna and Labarta, 2014).
Here is part of Sam’s pedigree. PCR and Southern blot analysis was used to elucidate the number of CGG repeats in various family members. These data reveal that Sam and his cousin each have more than the 200+ copies necessary to cause FXS.
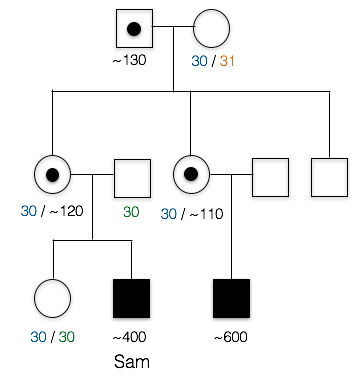
5. Based on these data, would Sam’s sister be expected to have FXS or pass on the condition?
No, she neither has FXS nor would she be expected to pass on the condition.
Both copies of Sam’s sister’s FMR1 genes have around 30 CGG repeats. This is considered to be within the normal range, so she would not have FXS. These repeats could expand slightly as she passed the genes on, but this expansion would usually only result in the premutation, not the full mutant FXS allele.
This pedigree also illustrates the point that (CGG)n premutations do not appear to expand when transmitted from father to daughter, but can expand (sometimes quite noticeably) into full mutations when passing through the female germline (Fragile X Syndrome, 2016; McMurray, 2010). There are several models for how this repeat expansion occurs. Recently, Zhao and Usdin (2015) have proposed that repeats can expand as a consequence of cellular DNA repair mechanisms.
6. Who is carrying a premutation in Sam’s family?
C. Sam’s mother, aunt, and grandfather carry the premutation.
A. incorrect. Several other individuals in the family have between 55 copies and 200 copies of the CGG repeat
B. incorrect. At least one other individual in the family has between 55 copies and 200 copies of the CGG repeat
D. incorrect. Sam’s first cousin has around 600 copies of the CGG repeat; this is considered a full mutation and not a premutation. Several individuals in the family do have between 55 copes and 200 copies of the CGG repeat, however.
FMR1 gene structure
The FMR1 gene has 17 exons, (Eichler, Richards, Gibbs, and Nelson, 1994). The diagram below shows the exons (numbered boxes), the transcription start site (bent arrow), the (CGG)n repeats (vertical lines in exon 1), and the translation start site (ATG) where protein synthesis will begin. See above for an interactive review of transcription and translation. Click here for a review of introns, exons, and splicing.
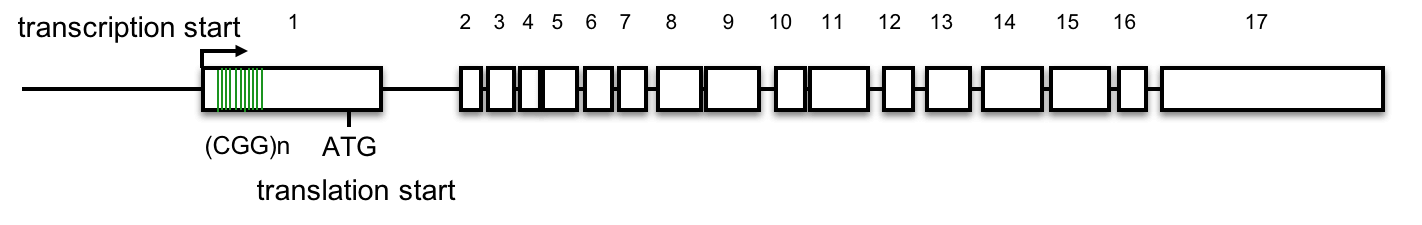
7. Based on this gene structure, how might (CGG)n expansion affect FMR gene expression or the FMR protein?
A. No, the (CGG)n repeats lie 5’ to the first ATG that begins translation. This lies outside the FMR1 open reading frame. Click here to learn more.
The most common mutations in the FMR1 gene linked to FXS result from the expansion of a (CGG)n triplet repeat in the 5’ untranslated region of the gene, within the first exon. (Beilina et al., 2004; Kremer et al., 1991).
Detailed analysis of the FMR1 gene delineates the (CGG)n repeat from -129 to -72, relative to the first in-frame ATG (Beilina et al., 2004), so the (CGG)n does not affect the open reading frame.
B. No, the (CGG)n repeats lie 5’ to the first ATG that begins translation. This lies outside the FMR1 open reading frame. Click here to learn more
The most common mutations in the FMR1 gene linked to FXS result from the expansion of a (CGG)n triplet repeat in the 5’ untranslated region of the gene, within the first exon. (Beilina et al., 2004; Kremer et al., 1991)
Detailed analysis of the FMR1 gene delineates the (CGG)n repeat from -129 to -72, relative to the first in-frame ATG (Beilina et al., 2004), so the (CGG)n does not affect the open reading frame.
C. Correct. Evidence suggests that FMR-1 expression levels are changed in individuals with FXS.
Epigenetic Silencing and FXS
Let’s look at some data. FMR1 mRNA levels from cells derived from two unaffected individuals or four FXS patients were quantified using reverse transcription followed by PCR, RT-PCR. Densitometric scans of the gels are shown in the six panels. The height of the peaks indicates the amount of each class of mRNA. The HPRT gene serves as a normalization control (control) in each experiment and is not expected to change appreciably.
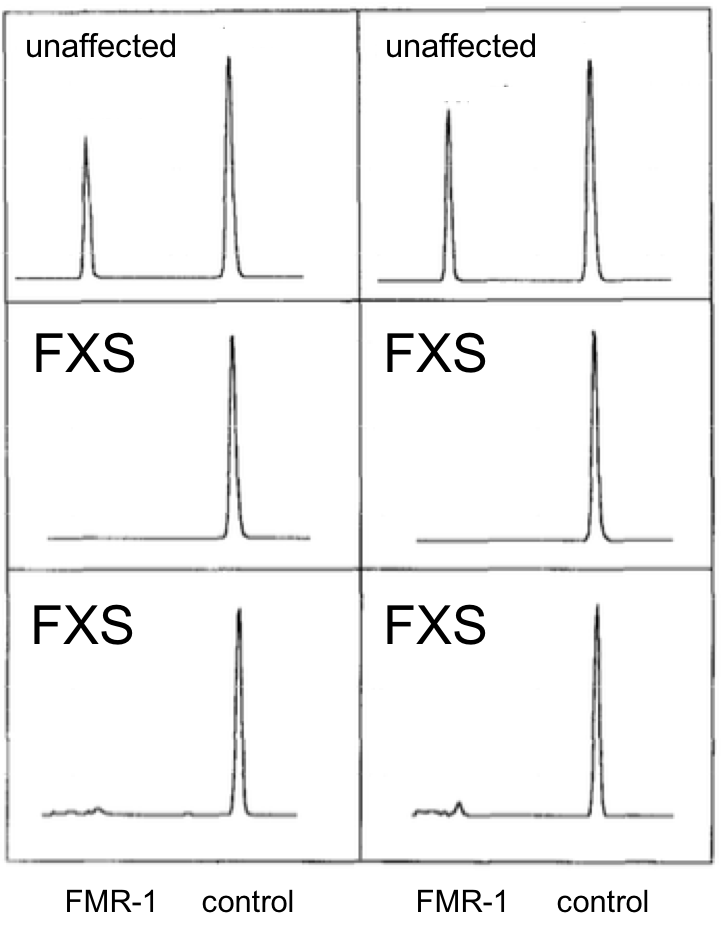
8. Based on these data, what can you conclude about the effect of the fragile X mutation on expression of the FMR1 gene?
B. Correct. FMR-1 expression is eliminated or almost eliminated in FXS patients.
A. incorrect. Compared to the FMR-1 peak in normal individuals, the expression peak for FXS is nonexistent (middle two panels) or drastically reduced (bottom two panels)
C. incorrect. Compared to the FMR-1 peak in normal individuals, the expression peak for FXS is nonexistent (middle two panels) or drastically reduced (bottom two panels)
Triplet repeats have been implicated in genetic disease phenotypes through a mechanism of epigenetic gene silencing. (Nageshwaran and Festenstein, 2015). How might this work? One possibility is DNA methylation. DNA methylation is a characteristic feature of heterochromatic, silenced regions of the genome. Sutcliffe et al., (1992) demonstrated increased methylation levels in the FMR-1 gene in FXS patients.
Click here for a refresher on DNA methylation.
The effects of DNA methylation on the expression of a typical gene are diagrammed below:

The data below from Brasa et al., (2016) shows the 5-methyl cytosine levels (5mC) in the region surrounding the FMR1 transcription start site (bent arrow) in three FXS patients, labeled A, B, and C. The 5mC levels in these patients are shown in red, while blue is the average 5mC levels in the corresponding region of four unaffected individuals used as controls.
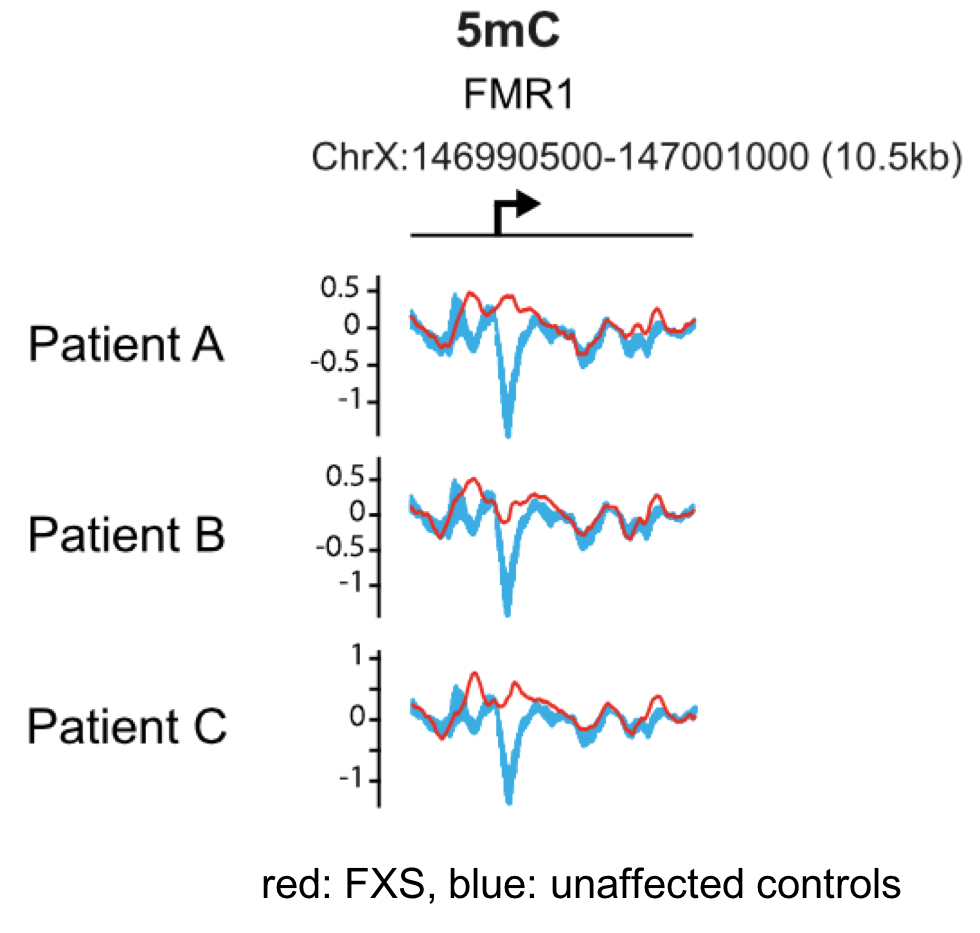
9. Based on the data, what can you conclude about DNA methylation in FXS patients?
B. Yes, the 5mC levels around the transcription start site are higher in FXS patients than in control individuals, and are high throughout this region of the gene. Current evidence suggests that the gene silencing mechanism involves DNA methylation and hydroxymethylation throughout the promoter region surrounding the (CGG)n repeat (Brasa et al., 2016).
A. incorrect. Note the dip in levels of 5mC centered around the transcription start site in control individuals (blue).
C. incorrect. Note the dip in levels of of 5mC centered around the transcription start site in control individuals (blue).
Other components important in epigenetic regulation, such as modifications to core histones, also appear to be important in the case of FMR-1. One such modification to histone 3 involves tri-methylation of lysine 9 (H3K9Me3). H3K9me3 is associated with heterochromatin and gene silencing (Becker, Nicetto, and Zaret, 2015).
Before examining the chromatin packaging state of FMR-1, these three links might be helpful review:
The data below, adapted from Kumari and Usdin (2010), summarize the results of chromatin immunoprecipitation (ChIP) assays for the presence of H3K9me3 across the FMR-1 gene in cell lines derived from two healthy individuals and four FXS patients. H3K9me3 is a characteristic feature of heterochromatin (Becker, Nicetto, and Zaret, 2015). The authors compared methylated histone levels in the first exon, which contains the (CGG)n repeat, to those within the first intron, as shown in A. Part B indicates the relative abundance of H3K9me3. All methylated histone levels across FMR-1 in the FXS patient-derived cells were statistically significant compared to those of the healthy controls. GAPDH serves as an internal control in the experiment. You may wish to click here for a humorous interpretation of how the ChIP assay is done.
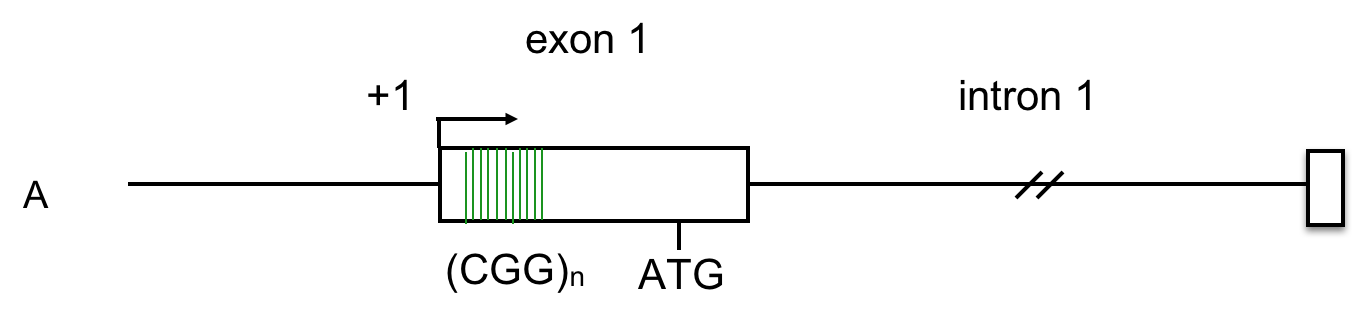
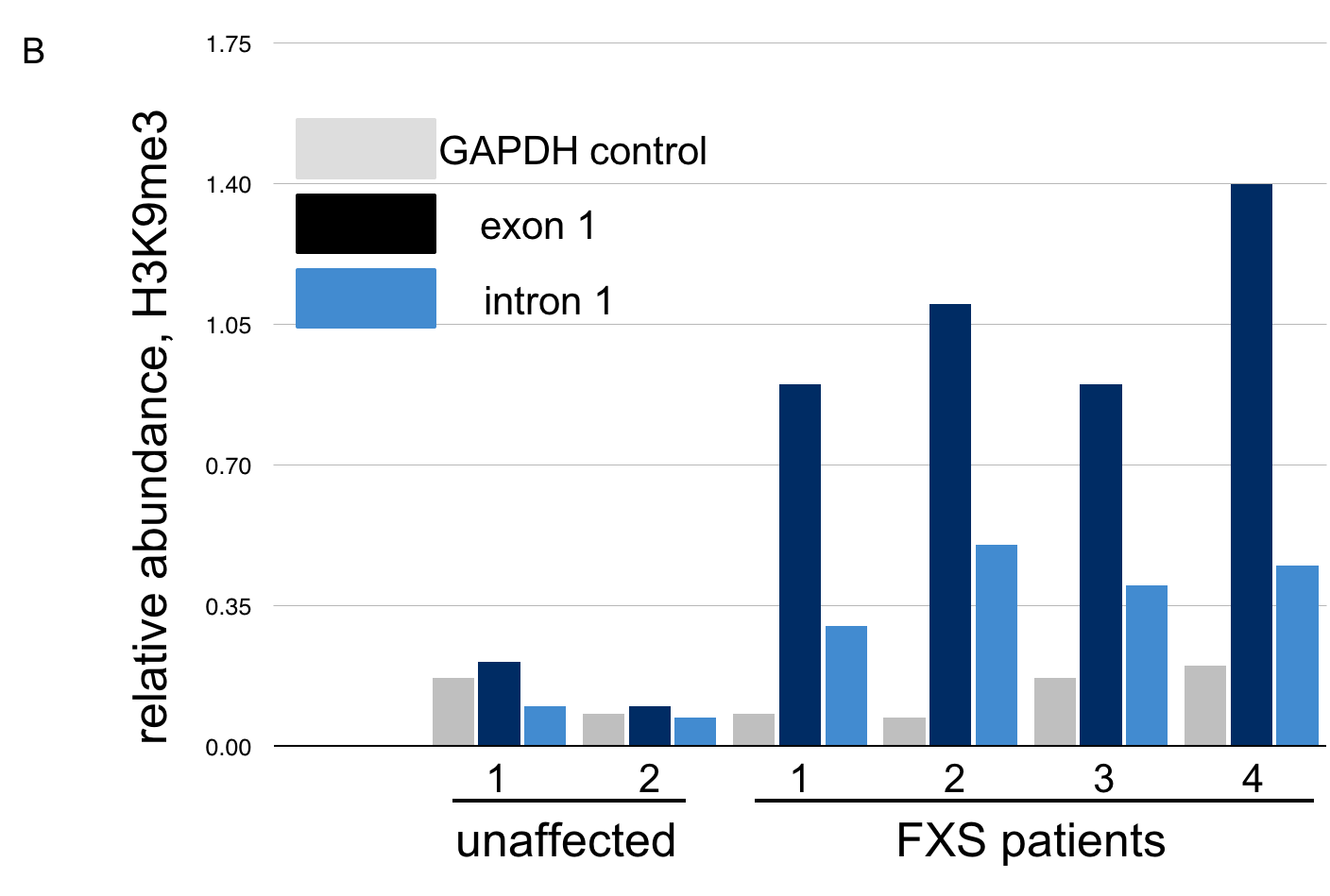
10. Based on these data, what can we conclude about the presence of repressive histone modifications on the FMR1 gene?
C. The levels of H3K9me3, characteristic of heterochromatin, are highest across FMR-1 exon 1 in FXS patients. These patients have low levels of FMR-1 expression. Click here to learn more about the chromatin features of FMR-1 in FXS patients.
Examination of histone methylation patterns shows heterochromatic marks, such as trimethylation of lysine 9 and/or lysine 27 of histone H3 (H3K9me3 and H3K27me3)(Kumari and Usdin, 2010). Additionally, the FMR1 gene displays histone hypoacetylation (Pietrobono et al., 2005). These features are characteristic of repressive chromatin domains. CpG methylation has not been demonstrated in all cases of FMR1 silencing, but repressive modifications of histones appear to be consistently associated with the mutation (Pietrobono et al., 2005; Tabolacci et al., 2008).
A. Incorrect. The FMR1 gene is active in healthy individuals. Note the low levels of H3K9me3, similar to the GAPDH control, in cell lines from these individuals.
B. Incorrect. The FMR1 gene is silenced in FXS patients. Note the high levels of repressive histone modification H3K9me3 in all FXS patients, both across exon 1 and intron 1.
Neuron morphology
FXS is a disorder that has profound effects on the function of the central nervous system (CNS). Neurons are the cellular units of the CNS. A neuron receives chemical signals through its dendrites and sends electrical signals down the axon to the axon terminal. Here, the electrical signal is converted into a new chemical signal that is sent to the next neuron’s dendrites.
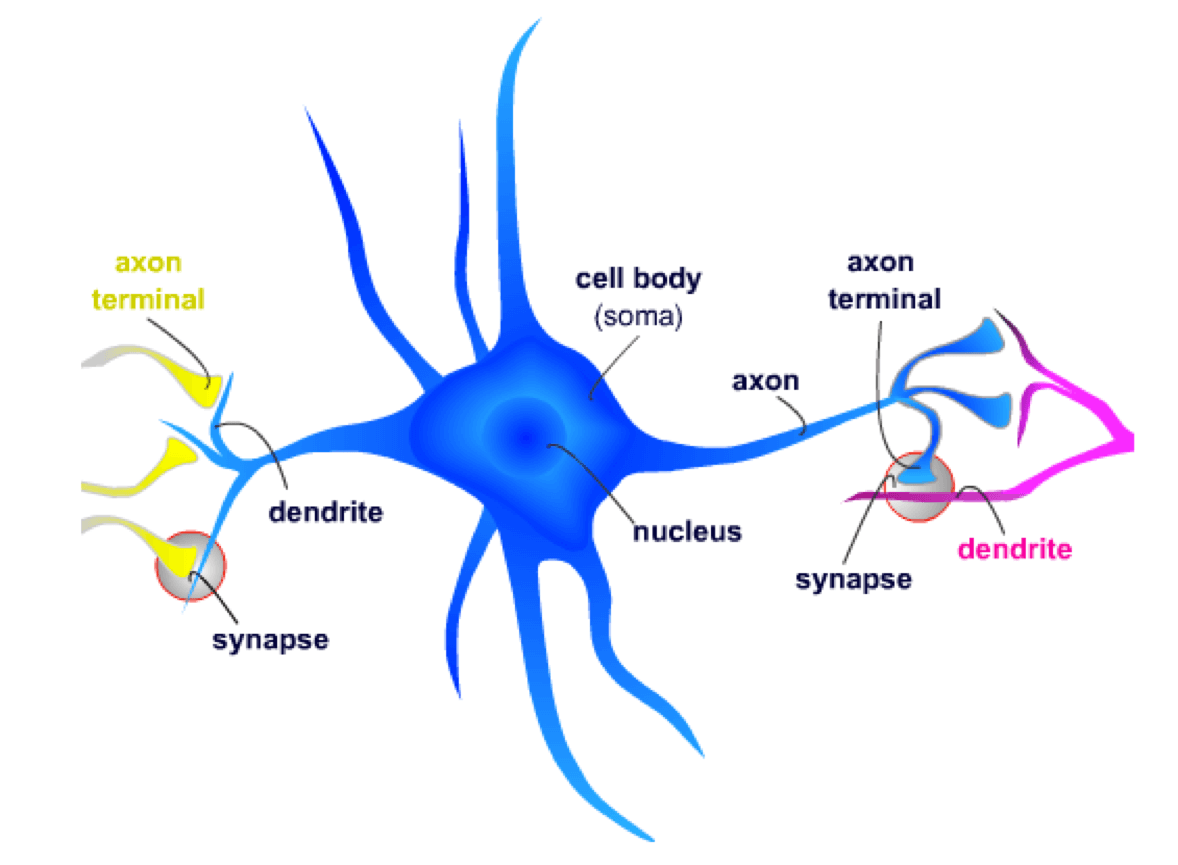
At the synapse, the axon terminal from the presynaptic neuron contacts a dendritic spine on the postsynaptic neuron. Neurotransmitters, which are small chemical signaling molecules, are released from the axon terminal and diffuse across the synaptic gap between the two neurons. After binding to receptors on the dendritic spines, a series of events is initiated that may result in the postsynaptic neuron signaling other neurons.

11. Each dendritic spine is a potential point of stimulation for the postsynaptic neuron. Which of the following would lead to an increase in the rate of postsynaptic neuron signaling?
B. correct. Since neurotransmitters stimulate the postsynaptic neuron, having more neurotransmitters in the synaptic gap would result in more receptor binding and greater signaling by the postsynaptic neuron.
A. incorrect. Fewer dendritic spines would mean fewer potential contact points for stimulation, so the postsynaptic neuron would send signals less often.
C. incorrect. Increasing the length of time between presynaptic signals would lower the amount of neurotransmitters in the synaptic gap, making the postsynaptic neuron less likely to fire.
Biological role of FMRP
Silencing of FMR1 appears to be the underlying cause of the FXS. Normal transcription of FMR1 produces the fragile X mental retardation protein (FMRP). FMRP is an RNA binding protein, expressed in the central nervous system and gonads (reviewed in Chen and Joseph, 2015). Lack of FMRP results in neurons having altered morphology and density of dendritic spines.
This figure shows dendrites from a FXS patient and an unaffected control individual. Note the profusion of dendritic spines in the person with FXS. Images represent extreme examples for illustration purposes.
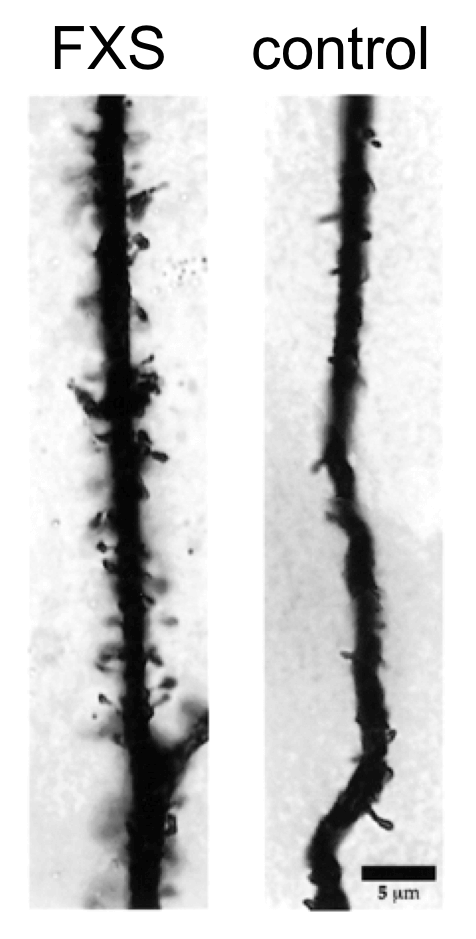
12. Based on the neuronal phenotype in FXS patients, which most likely represents the mode of action of FMRP on dendrite formation?
A. Correct. If repressing the production of proteins is the normal function of FMRP, then a lack of FMRP would be expected to increase the number of dendritic spines.
B. Incorrect. If FMRP stimulated the amount of proteins in dendritic spines, then we would expect a lack of FMRP to reduce the amount of these proteins.
C. Incorrect. FMRP binds to RNAs, while most transcription factors bind to the DNA in order to stimulate transcription.
FMRP’s normal role appears to be RNA export from the nucleus. RNAs that are bound by FMRP are not actively translated, so a lack of FMRP may result in unregulated translation of mRNAs.
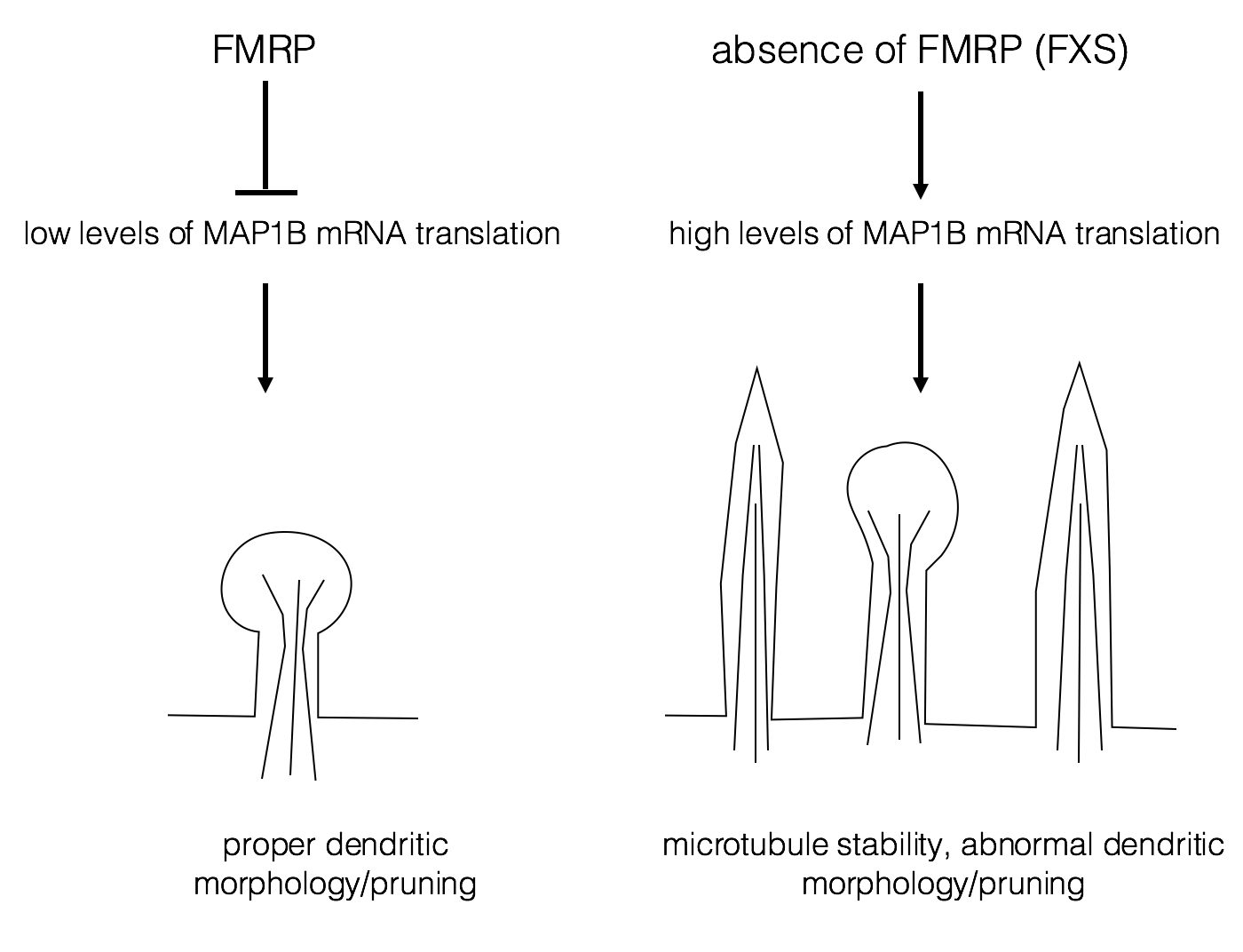
One possible target for FMRP is Map1B mRNA, whose protein product is important for dendrite development and synapse formation. (Lu et al., 2004; Zhang et al., 2001). FMRP normally represses translation of Map1B RNA. With normal levels of Map1B, the dendrites are easily pruned and remodeled as they mature. Pruning is a normal process of development in which excessive interneuronal contacts are removed (Luo and O’Leary, 2005). Excessive dendritic spines are often associated with autism (Tang et al., 2014). Lack of FMRP results in excess Map1B protein, leading to misshapen and excessive numbers of dendrites This may result in hyper-excitability of the neuron and some of the behavioral phenotypes of FXS.
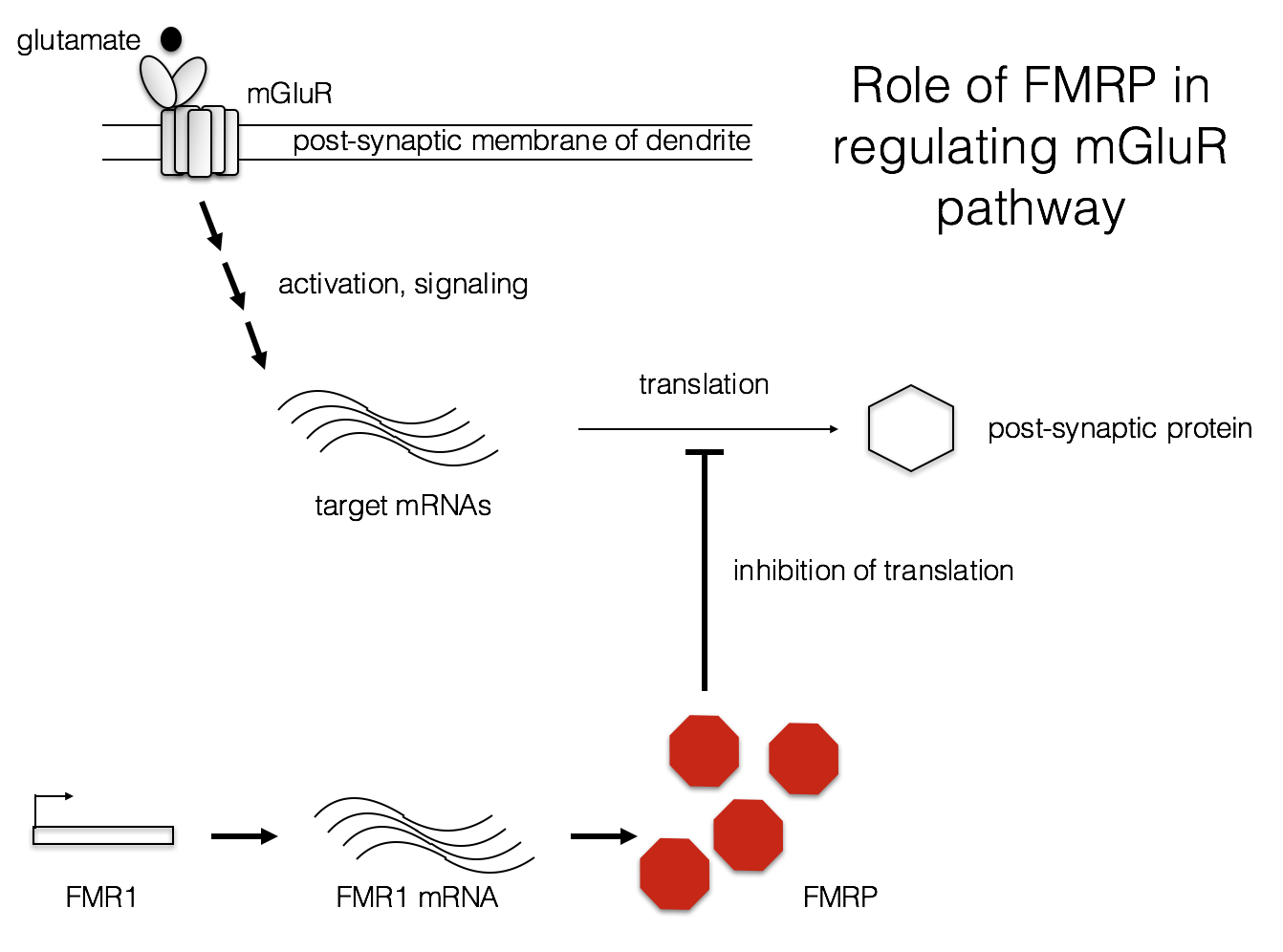
Another possible factor in the FXS phenotype is aberrant protein synthesis at the synapse. L-glutamate is one of the most common neurotransmitters in the CNS. Metabotropic glutamate receptors (mGluRs) bind L-glutamate and modulate a variety of responses in post-synaptic neurons (Niswender and Conn, 2010). In FXS, it may be that protein synthesis is abnormally high following mGluR activation (Bear, Huber, and Warren, 2004). Some of these protein products may alter the electrophysiology of these neurons, contributing to the FXS phenotype.
Possible therapies for FXS
Conventional therapies for the neurological effects of FXS
Recently, work from Deng and Klyachko (2016) suggests that increased mGluR5 signaling results in excessive sodium currents, lowered action potential threshold, and hyper-excitability of neurons in FXS. Targeting sodium current currents may help reduce the hyperexcitability observed in FXS patients.
Drugs that antagonize the mGluR and GABA signaling pathways (also reduced in FXS) are currently the focus of research into treatment for FXS patients (reviewed in Chen and Joseph, 2015). For more information on current research and clinical trials, visit the National Fragile X Foundation.
Epigenetic therapies for FXS
Drugs that inhibit DNA methylation, such as 5-azadC, have been tested on cells derived from FXS patients (Chiurazzi et al., 1998). Such treatment restored FMR1 activity in these cells, confirming the epigenetic nature of the FMR-1 silencing observed in FXS patients. However, methyltransferase inhibitors probably could not be used for long-term treatment of children due to toxic effects. They may, however, be useful in treating other FMR-1 related disorders such as FXTAS, which occur in older men (FXTAS, 2015).
Epigenetic therapy has moved to clinical trials in the treatment of Friedreich’s ataxia (Newman, 2015). As described above, Friedreich’s ataxia is another disease caused by trinucleotide repeat expansion and subsequent gene silencing by heterochromatin (Suhl and Warren, 2015). In this case, silencing of the frataxin gene may be reversible by the administration of vitamin B3 (nicotinamide) to patients. Nicotinamide inhibits histone deacetylases which contribute to heterochromatin formation (Newman, 2015).
Acknowledgments
We are indebted to M.I. and the Fragile X Resource Center of Missouri for their inspiration and assistance. Howard Granok wishes to thank the members of the Genomics Education Partnership, Sarah C. R. Elgin, and the members of the Elgin lab for comments on the original module and the resulting website. Yating Liu created the online version of the module, with assistance from Wilson Leung. This work was supported by NSF grant IUSE 1431407 awarded to Washington University on behalf of Sarah C. R. Elgin.
Resources
- Chromatin (2016). Retrieved from modENCODE website: http://modencode.sciencemag.org/chromatin/introduction
- Claire, C. (2014, May 3). Chromatin Immunoprecipitation. Retrieved September 25th, 2016 from Youtube: https://www.youtube.com/watch?v=YSZTvbcGaTc
- DNA methylation (nd). Retrieved from Cold Spring Harbor Laboratory DNA learning center: https://www.dnalc.org/view/17011-DNA-Methylation.html
- Epigenetics (2016). Retrieved from University of Utah Health Sciences website: http://learn.genetics.utah.edu/content/epigenetics/
- Histone methylation (nd). Retrieved from Cold Spring Harbor Laboratory DNA learning center: https://www.dnalc.org/view/17012--Histone-Methylation.html
- RT-PCR (2002). Retrieved from Davidson College website: http://www.bio.davidson.edu/people/kabernd/seminar/2002/method/lowry/rtpcr.htm
- Transcribe and Translate a Gene (2016). Retrieved from University of Utah Health Sciences website: http://learn.genetics.utah.edu/content/basics/transcribe/
- Transcription and Translation: mRNA splicing (nd). Retrieved from Cold Spring Harbor Laboratory DNA learning center: https://www.dnalc.org/resources/3d/24-mrna-splicing.html
- What is epigenetics? (nd). Retrieved from Cold Spring Harbor Laboratory DNA learning center: https://www.dnalc.org/view/17010-What-is-Epigenetics-.html
References
- Bear MF, Huber KM, Warren ST. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27:370–377.
- Becker, J.S., Nicetto, D., and Zaret, K.S. (2015). H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 32(1):29-41. doi: 10.1016/j.tig.2015.11.001.
- Beilina, A., Tassone, F, Schwartz, P.H., Sahota, P., and Hagerman, P. J. (2004). Redistribution of transcription start sites within the FMR1 promoter region with expansion of the downstream CGG-repeat element. Hum. Mol. Genet. 13 (5): 543-549. doi: 10.1093/hmg/ddh053
- Brasa, S. et al., (2016). Reciprocal changes in DNA methylation and hydroxymethylation and a broad repressive epigenetic switch characterize FMR1 transcriptional silencing in fragile X syndrome. Clin. Epigenetics 8: 15. doi: 10.1186/s13148-016-0181-x.
- Chen, E., and Joseph, S. (2015). Fragile X Mental Retardation Protein: A Paradigm for Translational Control by RNA-Binding Proteins. Biochimie.114: 147-154. doi: 10.1016/j.biochi.2015.02.005
- Chiurazzi, P., Pomponi, M.G., Willemsen, R., Oostra, B.A., and Neri, G. (1998). In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum Mol Genet. 7(1):109–113.
- Chromatin (2016). Retrieved from modENCODE website: http://modencode.sciencemag.org/chromatin/introduction
- Claire, C. (2014, May 3). Chromatin Immunoprecipitation. Retrieved September 25th, 2016 from Youtube: https://www.youtube.com/watch?v=YSZTvbcGaTc
- Deng, P-Y, and Klyachko, V.A. (2016). Increased Persistent Sodium Current Causes Neuronal Hyperexcitability in the Entorhinal Cortex of Fmr1 Knockout Mice. Cell Reports 6 (12): 3157–3166
- Dewald, G.W., Buckley, D.D., Spurbeck, J.L., and Jalal, S.M. (1992). Cytogenetic guidelines for fragile X studies tested in routine practice. Am J Med Genet. 44(6): 816-21.
- DNA methylation (nd). Retrieved from Cold Spring Harbor Laboratory DNA learning center:https://www.dnalc.org/view/17011-DNA-Methylation.html
- Eichler, E.E., Richards, S., Gibbs, R.A, and Nelson, D.L. (1994). Fine structure of the human FMR1 gene. Hum. Mol. Genet. 3(4): 684-685.
- Epigenetics (2016). Retrieved from University of Utah Health Sciences website: http://learn.genetics.utah.edu/content/epigenetics/
- Fortuño, C., and Labarta, E. (2014). Genetics of primary ovarian insufficiency: a review. J Assist Reprod Genet. 31(12): 1573–1585. doi: 10.1007/s10815-014-0342-9
- Fragile X Sydrome (2016, July 5). Retrieved from NIH Genetics Home Reference website: https://ghr.nlm.nih.gov/condition/fragile-x-syndrome#statistics
- FXTAS (2015). Retrieved from National Fragile X Foundation website: https://fragilex.org/fragile-x/fxtas/
- Hagerman R, Hagerman P. (2013). Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol 12 (8):786–798.
- Hecht F., Sutherland, G.R. (1985). Detection of fragile sites on human chromosomes. Clin. Genet. 28:95–96
- Histone methylation (nd). Retrieved from Cold Spring Harbor Laboratory DNA learning center: https://www.dnalc.org/view/17012--Histone-Methylation.html
- Irwin, S.A., Galvez, R., and Greenough, W.T. (2000) Dendritic spine structural abnormalities in Fragile X mental retardation syndrome. Cerebral Cortex 10: 1038-1044.
- Kremer E, Pritchard M, Lynch M, Yu S, Holman K, Baker E, et al. (1991) Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252 (5013):1711–1714
- Kumari D, and Usdin K. (2010). The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in fragile X syndrome. Hum Mol Genet 19(23):4634–4642.
- Luo L., O'Leary D.D. (2005). Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 28:127–56.
- Lu, R., et al., (2004). The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc. Natl. Acad. Sci. USA 101(42): 15201-15206.
- McMurray, C.T. (2010). Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 11: 786–799. doi: 10.1038/nrg2828
- Nageshwaran, S. and Festenstein, R. (2015). Epigenetics and Triplet-Repeat Neurological Diseases. Front Neurol. 6: 262. doi: 10.3389/fneur.2015.00262.
- Newman, M (2015, January 28). Friedreich’s Ataxia Treated With Vitamin B3 in Clinical Trial. Retrieved from Friedreich’s Ataxia News website: http://friedreichsataxianews.com/2015/01/28/friedreichs-ataxia-treated-with-vitamin-b3-in-clinical-trial/
- Niswender, C.M. and Conn, J.P. (2010). Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacol. Toxicol. 50: 295-322. doi: 10.1146/annurev.pharmtox.011008.145533
- Pieretti, M., et al. (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66: 817-822.
- Pietrobono, R., et al., (2005). Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum. Mol. Genet. 14(2): 267-77. doi: 10.1093/hmg/ ddi024.
- RT-PCR (2002). Retrieved from Davidson College website: http://www.bio.davidson.edu/people/kabernd/seminar/2002/method/lowry/rtpcr.htm
- Santoro M.R., Bray S.M., and Warren, S.T. (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 7:219-45. doi: 10.1146/annurev-pathol-011811-132457.
- Schwartz, M., Zlotorynski, E., and Kerem, B. (2006). The molecular basis of common and rare fragile sites. Cancer Letters 232 (1): 13-26.
- Stufflebeam, R. (2008). Neurons, synapses, action potentials, and neurotransmission. Retrieved from CCSI website: http://www.mind.ilstu.edu/curriculum/neurons_intro/neurons_intro.php
- Suhl, J.A., and Warren, S. T (2015). Single-Nucleotide Mutations in FMR1 Reveal Novel Functions and Regulatory Mechanisms of the Fragile X Syndrome Protein FMRP. J Exp Neurosci. 9( Suppl 2): 35–41. Published online 2015 Dec 8. doi: 10.4137/JEN.S25524
- Sutcliffe, J.S., et al. (1992). DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1 (6): 397-400.
- Sutherland, G.R. (1977). Fragile sites on human chromosomes: demonstration of their dependence on the type of tissue culture medium. Science 197: 265–66.
- Tabolacci E, Moscato U, Zalfa F, Bagni C, Chiurazzi P, Neri G (2008). Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur J Hum Genet.16(12):1487-1498. doi: 10.1038/ejhg.2008.130.
- Tang, G., et al. (2014). Loss of motor-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83:1131–1143.
- Transcribe and Translate a Gene (2016). Retrieved from University of Utah Health Sciences website: http://learn.genetics.utah.edu/content/basics/transcribe/
- Transcription and Translation: mRNA splicing (nd). Retrieved from Cold Spring Harbor Laboratory DNA learning center: https://www.dnalc.org/resources/3d/24-mrna-splicing.html
- Verkerk, A.J.M.H., Pieretti, M., Sutcliffe, J.S., Fu, Y-H, Kuhl, D.P.A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–14.
- Warren, S. T., and Nelson, D.L. (1994). Advances in Molecular Analysis of Fragile X Syndrome. JAMA. 271(7):536-542. doi:10.1001/jama.1994.03510310066040.
- What is epigenetics? (nd). Retrieved from Cold Spring Harbor Laboratory DNA learning center: https://www.dnalc.org/view/17010-What-is-Epigenetics-.html
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. (2001). Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107:591–603.
- Zhao, X-N., and Usdin, K. (2015). The Repeat Expansion Diseases: The dark side of DNA repair. DNA Repair 32: 96-105.